Since 2006, the platform carried out modelling and structure-based design studies for the benefit of chemical biology projects. From initial designs to the scientific publishing, including significative involvment in the supervision (in silico and structural biology parts) of 15 PhD programs.
About 30 molecular types were covered, such as: modified DNAs, enzymes related with glycolysis, enzyme and carriers of lipid signaling pathways, superoxide dismutase, carbonic anhydrase, checkpoint kinases, histone acetyl transferase, serpins, nuclear receptors, enzymes of mycolic acids biosynthesis, DNA repair protein, indoleamine dioxygenase, hydroxysteroid dehydrogenase, nitric oxide synthase ...
The way we work
The level of available information found in structural databases is massively rising, even if this knowledge is not adapted to direct batch processing in the field of structure-based drug design. Finally, the databases provide structural waypoints that we may consider as independent or correlated snapshots of conformal motions (inherent, induced fit ...) related to different events and mechanisms. In these cases, the problems to solve are many and different from those encountered in most screening approaches.
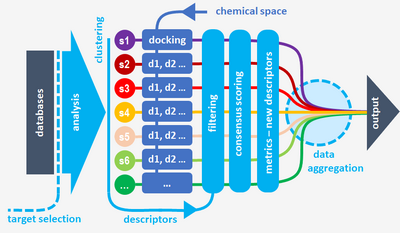
The structures used to sample a given protein (enzyme, carrier, receptor …) may not respond in the same way to a given docking protocol (including searching, scoring, clustering scheme and post-docking steps) relatively to a chemical (ligands) space. In order to obtain results as unbiased as possible, consensus methods using multiple docking protocols and multiple scoring functions, may be used. Consequently, the analyses may take into account new descriptors such as similarity, efficiency, stability … in addition to the classical ligand-protein affinity quantities.
All of these elements should be considered as components of multimodal docking protocols based on consensus scoring, similarity filtering, cross docking methods, including optimizations (pharmacophore models, GPU, flexibility) of searching (docking) phase. This means that structural analyzes driven before docking, descriptor’s based post-docking filters, in silico efficiency metrics, data aggregation methods, full automation of multiple software are becoming critical issues in order to calculate (sometimes) less but better.
Contact
frederic.rodriguez@univ-tlse3.fr
ResearchGate

BuildBlog
